
New Delhi: The PathGennie algorithm was developed to simulate rare molecular events in drug discovery.
Scientists at the S. N. Bose National Centre for Basic Sciences, Kolkata, created the new computational framework to address a major limitation in computer-aided drug discovery. Published in the Journal of Chemical Theory and Computation, the open-source software predicted how drug molecules unbound from protein targets while preserving true physical pathways.
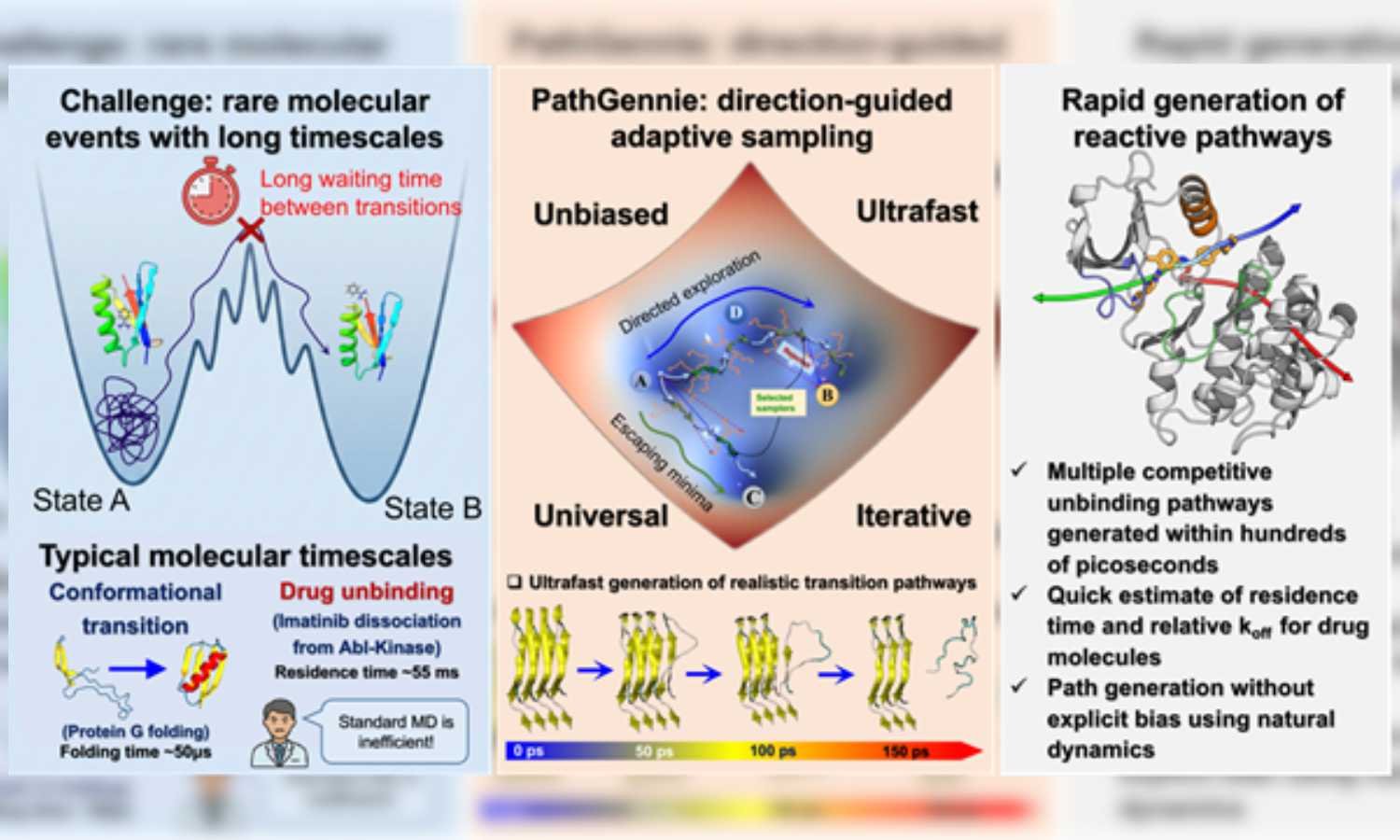
Understanding how long a drug remained attached to its target protein, known as residence time, often played a greater role in effectiveness than binding strength alone. However, simulating drug unbinding was difficult because these rare molecular events occurred over milliseconds to seconds. Standard molecular dynamics simulations struggled to reach such time scales.
As a workaround, researchers traditionally applied artificial bias forces or elevated temperatures to force unbinding. Although effective in speeding up simulations, these methods distorted molecular physics and produced unreliable transition pathways. The new approach avoided these distortions by keeping simulations fully unbiased.
How the PathGennie algorithm enables unbiased molecular simulations
Researchers designed the PathGennie algorithm to mimic natural selection at a microscopic level. Instead of forcing molecular motion, it launched swarms of ultrashort and unbiased simulation trajectories, each lasting only a few femtoseconds. Only trajectories that moved closer to a defined end state were selectively extended.
This direction-guided adaptive sampling allowed productive trajectories to survive while unproductive ones were discarded early. As a result, the method bypassed long waiting times linked to rare events and retained accurate kinetic pathways. The framework also operated across any collective variables, including high-dimensional or machine-learned coordinate spaces.
In proof-of-concept studies, a team led by Prof. Suman Chakrabarty, along with Dibyendu Maity and Shaheerah Shahid, tested the method on complex systems. The simulations mapped how a benzene molecule exited the deep binding pocket of the T4 lysozyme enzyme. The method also identified three distinct dissociation pathways for the anti-cancer drug imatinib as it unbound from the Abl kinase. These results matched earlier biased simulations and experimental findings.
Researchers said the PathGennie algorithm can be applied beyond drug discovery.
Because the framework was general in scope, scientists noted its relevance to chemical reactions, catalytic processes, phase transitions, and self-assembly phenomena. The software was released freely, allowing wider adoption across scientific communities and integration with modern machine-learning tools.